芳香族高分子系の電子移動反応
過渡吸収分光及び過渡二色性の測定によるホッピング過程の直接的測定と超長寿命電荷分離状態の作成
1. はじめに
側鎖に大きな芳香族基を持つビニルポリマー(下図)は、光照射によって電導性を示す有機物質(有機光電導体)の一つであり、ポリ(N-ビニルカルバゾール)(PVCz)のように、実際に光電導材料に使用されたものもある。これらの有機光電導体は、実用面からも多くの研究がなされており1-5)レーザーと物質の相互作用を利用した新規機能性の開発も試みられている6) 。実用的な材料としては、これらのポリマーより優れた物性を持つ分子系も現在では多く存在する。しかし一方これらのポリマー系に対しては巨視的な光電導挙動や分子レベルでの基礎物性に関する知見も多く蓄積されており、新たな機能性材料の設計指針を得るための参照物質と位置付けられている。また巨視的な材料物性と微視的な光化学過程との対応を検討し、機能発現機構を分子レベルから構築することは基礎的な光化学の研究対象としても重要と考えられる。また、これらの高分子は光励起によって電荷分離状態を生成することによって、電荷を長距離まで輸送することができる分子であり、植物の光合成反応中心のような光電変換分子と見なすことができる。これらの高分子系の電子移動過程を基礎的な観点から明らかにし、その知見に基き、新たな光電変換分子系の設計指針を提出することも、重要な課題である。
代表的な芳香族ビニルポリマーの構造
一般に芳香族ビニルポリマーは側鎖の芳香族基のカチオン状態を電荷担体としたホール電導を行う。このカチオンは、分光データからは1つもしくはせいぜい数個の芳香族基に局在化した状態として存在すると考えられている。したがって、これらの高分子系での光電導は、バンド理論よりはむしろ分子間電子移動反応を基本とする側鎖芳香族基間の電荷ホッピングとして取り扱いうる問題と考えられる。
一般に電子移動反応は、多くの化学反応に深く関わっており、理論・実験ともに数多くの研究がなされてきた7-11)。これらの結果に基づくと、芳香族ビニルポリマーの光電導初期過程は以下のような電子移動素過程の組み合わせとして記述できる。なおAは電子受容性ゲスト分子Dは芳香族基である。
---DDADDD-- → DDA-D+DD-- 電荷分離
---DDA-D+DD-- → DDADDD-- 電荷再結合
---DDA-D+DD-- → DDA- D D+D-- 電荷シフト
第一段階のキャリア発生過程は、①光励起によって生成した励起分子(芳香族基)がイオン化あるいは固体中にドープされた他分子との電子移動反応により電荷分離状態を生成する過程に対応し、第2段階のキャリア輸送過程は、②カチオン状態が近接の中性芳香族基に、次々と電荷シフト反応(ホッピング)を行う過程となる。光照射によって引き起こされる電荷分離の後の過程は、再結合過程と電荷シフト過程の競争過程である。したがってキャリア発生効率の向上のためにも、速い電荷シフト反応過程を持つことが望ましい。また言うまでもなく、電荷シフト反応ではキャリア輸送速度にも直接関連しているキープロセスである。
この電荷シフト反応の直接的な解明のためにレーザーを光源とした時間分解測定法による研究も多く行われてきた12-14) 。パルスレーザーを用いた過渡吸収分光法は、光誘起諸過程の研究に有効な手法として利用されている。しかし同種分子間で電荷シフト反応が起こっても、イオン種のスペクトル形状にはほとんど変化が現れないため、この電荷シフト過程を直接抽出するのは困難であった。
我々は、この電荷シフト反応の直接的な検出のために、レーザー光の偏光特性と短時間パルス性を利用した過渡吸収二色性の測定が有効であることを示し、この手法を種々の芳香族ビニルポリマーにおける光電導関連初期過程の解明に利用してきた。 ここではパルスレーザーを用いた我々の研究を中心に紹介し、分子レベルでの反応機構や電子移動反応理論からの考察を行う。またこれら高分子を用いた超長寿命電荷分離状態の生成についても言及する。
2. ポリビニルカルバゾールフィルム系の光電導初期過程
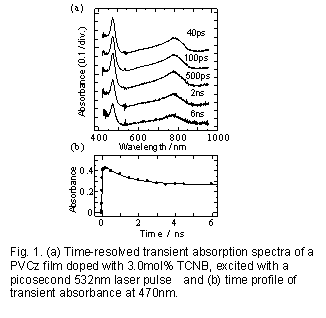
Fig.1(a)に電子受容体としてテトラシアノベンゼン(TCNB)を3モル%含むPVCz固体フィルムをピコ秒532nmレーザーで励起し、得られた過渡吸収スペクトルを示す15,16)。基底状態で生成したPVCz-TCNBの弱い電荷移動(CT)錯体が532nmで選択励起され電荷分離が起こり、TCNB-(470nm)とカルバゾールのカチオン状態(Cz+) (790nm付近)の吸収が励起直後から現れる。電荷分離状態の吸光度の時間変化をFig.1(b)に示した。この吸収は時定数1.26 nsで減衰しこの観測時間領域では約60%の吸収が一定成分として残る。Scheme 1のように生成したイオン対状態が、再結合(CR)過程と競争しホール移動(HT)過程を行い、数ナノ秒以降の吸収はCRを逃れたイオン状態によるとして解析した結果(Fig.1(b)の実線)は、観測値をよく再現した。
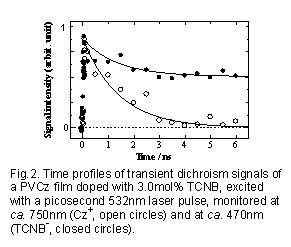
より直接的にホール移動過程を検出するために、イオン種の過渡吸収二色性の時間変化を検討した(Fig.2)。
Cz+の過渡吸収二色性(白丸)は、Fig.1で示した初期イオン対の寿命とほぼ同じ約1.2nsの時定数で0レベルまで減衰した。
CT吸収帯の偏光励起によって、生成直後のイオン対は偏光メモリーを持つが、固体フィルム中にランダムに存在する他のCz基へのホール移動が起こりCz+は存在するがその過渡吸収二色性は消失している。一方、TCNBの濃度は3mol%と低いため、この時間域ではアニオンは電荷シフト反応を行なわず、過渡二色性の時間依存性(黒丸)は過渡吸収と同じ挙動を示した。この測定によりPVCz高分子固体系の光電導初期過程が、第一近似的には比較的簡単なScheme1で現されることが明らかになった。他のカルバゾール系低分子アモルファス固体系でも同様のスキームが直接確認されている17)。
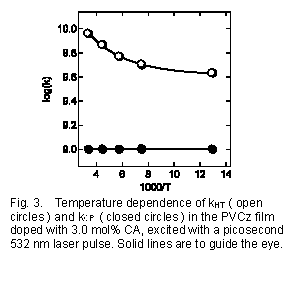
ホール移動速度及び電荷再結合速度の温度依存性も検討した18)。この解析のためには、Scheme1を三次元的に拡張したモデルが用いられているが、基本的には同じである。Fig.3に示すように、電荷再結合の速度定数は温度の低下とともに減少するが、低温ではあまり大きな温度依存性を示さない。多くの系の電荷再結合に対する温度依存性は、一般的な無輻射過程同様に、温度に依存する項(strong-coupling)及び温度に依存しない項、(weak-coupling)の2つで記述できる場合が多い7-11)。PVCzフィルム系における再結合過程も定性的には、通常の電荷再結合挙動と同様の温度依存性と考えられる。一方、室温から77Kの範囲ではホール移動速度定数は全く温度依存性を示さなかった。PVCz固体系のホール移動に対する理論的な計算19)からは、ホール移動反応の電子的な相互作用の大きさは側鎖Cz基の配向や距離に大きく依存することが示されている。これらの結果をあわせて考えると、実質的には活性化エネルギーを持たないような側鎖の小さな運動によってCz基の距離や配向が揺らぎ、その結果電子的な相互作用が増大する過程がホール移動に重要な役割を果たしていることが示唆される。
一般に、光電流測定からはPVCzのような固体高分子フィルムでのホール易動度は、温度の低下とともに低下することが知られている。一方、分子レベルでの電荷シフト反応はここに示したように少なくとも77Kまでは温度依存性は示さなかった。お互い相矛盾するように思われるが、光電流は長い距離と長い時間を経て起こる過程の結果であり、主にカチオンが比較的安定なサイトにトラップされた後の活性化過程を要する脱トラップ過程に律速されている。このような安定なトラップサイトは、Cz単位として1000個に数個程度であると見積もられている。この実験のような比較的短い時間領域では、未だトラップされていないカチオン種を観測しているため、温度効果が観測されなかったと考えられる。事実、数百ナノ秒以降の時間領域では、過渡吸収の時間変化に温度効果が観測されている。
3.従来の光電導メカニズムとの比較
一般にこれら高分子固体系の誘電率は3から4程度であり、低極性環境である。そのため電荷分離後、隣接したCz基へのホール移動にはアニオンからのクーロン引力に逆らう0.3から1eV以上の大きな障壁が存在する。このような障壁があるにも関わらずホール移動が行われる理由として、生成直後の電荷分離状態が熱化過程の間に余剰エネルギー利用し、距離r0離れたイオン対を形成しクーロン引力を軽減するためと考えられてきた。この考え方は、初期熱化距離(r0)と外部電場の大きさを重要な因子とするOnsagerモデルとして、(光)有機導電体の研究では現在でも広く応用されている。PVCz系でこのr0は20から30Å程度と見積もられており、側鎖のCz基の数として5から10個程度に対応した電荷シフト過程が非常に高速に進行することを前提としている。一般に熱化過程は余剰振動エネルギーの散逸過程である。この振動緩和過程は、凝縮系では数ピコ秒からせいぜい数10ピコ秒といった短い時間領域で起こる。しかし二色性の測定結果のように、ホール移動過程は、数ピコ秒から10ピコ秒オーダーで起こるような振動緩和と競争する迅速なものではなく、ナノ秒領域で再結合過程と競争する比較的ゆっくりとした過程である事が直接示された。
このように従来のOnsagerモデルは、分子レベルでの実際の反応素過程の観点からいえば誤ったスキームに基づいている。しかし一方、このモデルが多くの光電導現象の幅広い実験結果を説明できるのも事実である。このOnsagerモデルに現れる初期熱化距離を、実際の分子スケールでのダイナミックスの点から合理的に解釈するヒントについては後述する。
我々の実験結果の提出後、単純に考えると吸熱過程であるにもかかわらず109s-1もの大きな速度定数を持つホール移動過程を合理的に説明するために、サイト間のエネルギー分布を持たせたモデル20)を基本に、モンテカルロシミュレーションによる検討も行われた21,22)。すなわち、サイトエネルギーの不均一分布が存在し、障壁を越える必要のないほぼ等エネルギーのサイトを経てホール移動が行われるとするメカニズムである。この結果からは、サイト間のエネルギー分布を0.3eVの標準偏差を持つガウス分布とした場合、実験値がよく再現できエネルギーサイト分布がホール移動過程に重要な役割を果たしていることも示されている。
また最近、TCNBを含むPVCzフィルムの電子移動ダイナミックスに対して、時間分解ESRを用いた測定も応用されている23)。スペクトルの解析から見積もられた励起後1マイクロ秒でのTCNB+とCz+は、8.4A程度の対間距離を持つことが報告されている。比較的近い対間距離をを持つ理由としては、アニオンに近いトラップサイトに補足されたCz+の存在が示唆されている。
4.溶液中での電子移動過程
溶液中の一般的な光誘起電子移動過程は理論や実験両面から多くの研究が行われており、これらの研究結果と高分子系の電子ダイナミックスを直接比較することによって、その特徴を知ることができる。
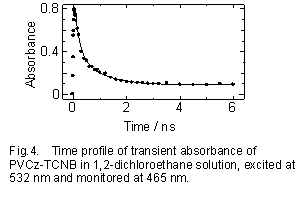
PVCz-TCNB系の1,2-ジクロロエタン溶液中における電荷分離状態の時間変化をFig.4に示す16,24)。溶液系でもフィルム系同様に基底状態で生成したPVCz-TCNBの弱い電荷移動錯体が532nmで選択励起され電状態の時間変化を荷分離し励起直後からTCNB-とCz+の吸収が現れ、サブナノ秒から数ナノ秒領域において非指数関数的に減衰する。固体フィルム系と異なり、溶液中では高分子鎖はそれぞれ独立に存在することを考慮に入れ、Scheme 2のようにAとD0の間で生成したカチオン状態が、再結合と競争し高分子鎖に沿ってホール移動過程を行い再びD0に戻ったときに再結合する電子移動ダイナミックスを提案した。
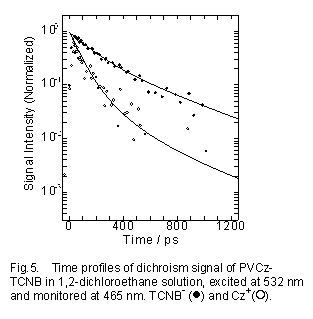
Fig.4の実線は再結合速度定数にはモノマーモデル系の値を用い、またホール移動速度定数kHTiは全て同じ値として(kHT=2.0×109s-1)計算した結果であり実験結果を再現する。イオン種の吸収の過渡吸収二色性の測定結果をFig.5に示す。溶液中では高分子鎖の運動(局所回転運動)によっても二色性の信号は消失するが、フィルム系と同様にカチオン状態の側鎖に沿った他のCz基へのホール移動も消失に寄与する。Fig.5の黒丸は、TCNB-の波長でモニターした結果である。この減衰はPopulation DecayであるP(t)と高分子鎖の局所回転(Micro-Browinian Motion)の時関数 R(t)の積、として表される。実線は、P(t)としてFig.4の計算結果、またR(t)の局所回転の時定数を950psとした計算で実験結果をよく再現している。
溶液中の高分子の局所緩和の時定数は、サブナノ秒から数ナノ秒と知られており、このR(t)はほぼ妥当な値といえる。一方、Cz+の過渡吸収二色性は(白丸)、アニオンのものより迅速に減衰している。過渡吸収は全てのCz+ (Di+)を観測しているが、二色性の信号は偏光励起メモリーが存在するCz+ (D0+) のみに由来する。したがって、D0+から他のDiにホール移動が起こったため、アニオンよりも速い減衰が観測されたと考えられる。実線はP(t)としてScheme2のD0+のみの時間変化を計算した結果である。計算には、Fig.4と同じ再結合速度定数及びホール移動速度定数、またR(t)は同じ950psの時定数を用いた。 この計算結果はカチオンの時間変化も良く再現しており、Scheme 2の妥当性が過渡吸収二色性の測定から確認された。
同様の手法によって1、2-ジクロロエタン溶液中での種々芳香族ビニルポリマーのホール移動速度定数が得られた16,26,27,28)。その結果、側鎖間の重なりが大きいと考えられる高分子系が、大きなホール移動速度定数を持つこと、また速度定数は、高分子の主鎖の局所緩和の時定数とは相関がなく、主鎖の運動よりも迅速にホール移動が進行する場合も多いことが明かとなった。
一般的には高分子系では側鎖間の重なりによって、エキシマーやダイマーカチオンが効率よく生成し、エキシマーは励起エネルギー移動の際の、またダイマーカチオンはホール移動の際のトラップサイトとして作用することが知られている。したがってこのような高分子系においてScheme 2のような1次元ランダムウォークモデルで電子移動ダイナミックスが記述できることは、効果的なトラップサイトが存在しないことに対応する。大きなホール移動速度定数を持つカルバゾール系高分子のモノマーモデル化合物であるエチルカルバゾールやエチルベンゾカルバゾールは、分子間でのダイマーカチオンやエキシマー生成の平衡定数が非常に小さいことが知られている。したがって高分子中でもあまり大きな安定化エネルギーを持たず、速いホール移動過程に有利に働いていると考えれられる。
またScheme 2では、D0+からD1+へのescape過程とその逆のホール移動過程を同じ速度定数としている。最初のホール移動に対するA-・D+D→ A-・DD+のモデル反応は、アニオン-カチオン間のクーロン引力に逆らって進行する過程であり、単純にクーロン引力の損失から考えるとジクロロエタンのような中極性溶媒中でも、10kT(kTは室温での熱振動のエネルギー)以上の吸熱反応となる。したがって電子移動反応のみならず一般の化学反応として考えても、この吸熱性の大きな反応が109/secもの大きな速度定数を持つことは単純には説明できない。
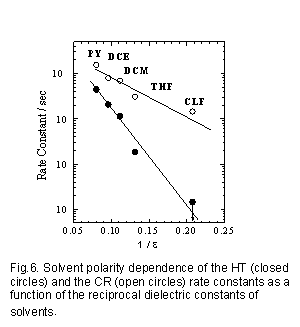
この理由を探るために、PVCz系の電子移動ダイナミックスをピリジン(Py)、1,2-ジクロロエタン(DCE)、ジクロロメタン(DCM)、THF、クロロフォルム(CLF)の5種の溶媒中で測定し溶媒効果を検討した28)。各々の溶媒中での電荷再結合速度定数(kCR)とホール移動速度定数(kHT)の溶媒誘電率に対する依存性をFig.6に示した。
再結合速度定数についてまず議論を行う。一般に基底状態で生成した弱いCT錯体を光励起し生成したイオン対の電荷再結合速度定数は、次のようなエネルギーギャップ依存性を示すことが知られている11,29,30)。(1) 電子移動反応としては逆転領域のみの振る舞いを示し、基底状態とイオン対状態のエネルギーギャップが小さくなるに従って反応速度が大きくなる。(2) このエネルギーギャップ依存性は溶媒に依存しない。ある溶媒中でDとAを変化させて得られるエネルギーギャップ依存性も、DとAを固定して溶媒を変化させて得られる場合とほぼ同じ依存性を示す事が知られている。単純にBornの式を用いた場合、同じDA系ではイオン対のエネルギーレベルは誘電率の逆数に依存する。今回観測されたPVCz系の再結合速度定数も、おおむねこれらのCT錯体を励起した場合に観測される挙動として理解できる。
一方、ホール移動速度定数も誘電率の低下とともに小さくなるが、その傾きは再結合速度定数と比較すると大きな値を持ちクロロフォルム溶媒中では実質的にはホール移動過程は観測されない。この結果は、確かにホール移動過程はクーロン引力に逆らう吸熱的な反応であり、溶媒の極性が減少するとともに実質的なクーロン引力が大きくなるために反応が起こりにくくなった事を示している。しかし、先述のようにジクロエタンやピリジンのような溶媒でも、モデル反応で考えられるエネルギー差は10kTもの大きな値を持つ。
このような本質的に速いホール移動が起こる理由に対しては、現在の所、カチオンがいくつかのCz基に非局在化した状態で存在する可能性を考えている。PVCz系のCzカチオンの吸収はモノマーモデルであるエチルカルバゾールのものと比較してブロードな形状を示し、他のCz基との相互作用の存在を示唆する。いくつかの芳香族基にカチオンの非局在化が起これば、実質的なアニオンからの距離が大きくなりクーロン引力が減少する。更に、溶質サイズが大きくなると溶媒の再配向エネルギーは小さくなるため、ホール移動に有効に作用する。この非局在化のサイズについては、未だ実験的な情報を得ることはできていない。しかし、土田らによるビニルカルバゾールオリゴマーにおけるカチオン状態の近赤外部の電荷共鳴吸収帯の測定によると31)、電荷共鳴帯は重合度の増大とともに20量体程度まで長波長へシフトする事が示され、数個程度のCz基に非局在化したカチオン状態が生成しそのサイズが熱的に揺らいでいることが示唆されている。ポリビニルピレンなどではこのような挙動は観測されておらず、特異的にPVCzでは非局在化と同定できる重合度依存性が観測されることが示されている。
完全にカチオンが非局在化してしまえば、過渡吸収の二色性も観測されないと考えれれる。従って土田らの指摘したように局在化したカチオン状態と非局在化したカチオン状態が、いわば速い平衡にあって小さな揺らぎによってこれらの状態を行き来していると考えられる。フィルム系のように低極性環境にも関わらず比較的迅速にホール移動が進行するのは、溶液中では高分子鎖はそれぞれ独立に存在するが、フィルム中ではCz基の密度が高く、近傍のCz基との相互作用によって非局在化ができる事が重要な因子になっていると考えるとよく説明ができる。またこのような非局在化した状態の実質的なサイズが、従来考えられてきたr0と関係していると考えている。実験的に、これらのサイズを見積もることが今後の課題であり、フェムト秒時間分解測定の結果から、このサイズに対する直接的な実験結果が得られつつある。
Return to top
5.固体吸着系におけるホール移動と超長寿命電荷分離状態の生成
植物の光合成反応中心のように光誘起電荷分離の後効率の良い電荷シフト反応を経て長寿命の電荷分離状態を人工的に作成しようとする試みは、数多く行われている31,32)。高分子鎖に沿ったホール移動によって増大したアニオン-カチオン対間距離を固定化した場合には、再結合速度が減少するため長寿命の電荷分離状態が生成することが期待できる。ここでは芳香族ビニルポリマーを用いた超長寿命電荷分離状態の生成について述べる。
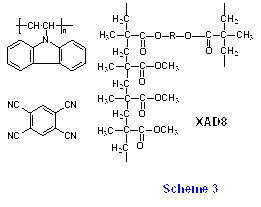
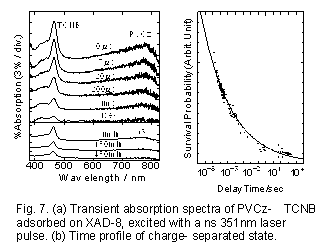
Fig.7(a)にPVCzとTCNBを多孔性吸着剤XAD-8に共吸着し(Scheme 3)、室温22℃でナノ秒351nmレーザー光照射した場合の過渡吸収スペクトルとその時間変化を示す33)。この図からわかるように、TCNB-及びCz+の吸収が励起後8時間においても観測された。まキセノンランプで同系を定常光照射した場合にも同様の超長寿命電荷分離状態が生成することから、レーザー多光子イオン化などによる非線形過程が関与しているわけではない。また長い時間領域での再結合挙動は77Kでもほとんど変化しないことから、長距離トンネリングによる再結合が起こっていると考えられる。
Fig.7(b)の実線は次のモデルに基づいて計算した結果である。まず電子受容体(A)に近接したカルバゾール基(D)の間で光誘起電荷分離の後、側鎖のDに沿ったホール移動が起こり対間距離が増大していく。それぞれのDは、XAD-8の表面の極性基(エステル基)によって、その周囲の極性環境がそれぞれ異なる。XAD-8のエステル基に結合したメチル基をバルキーなオクチル基に置換した場合には、このような長寿命電荷分離状態は生成しないことから、カチオンは高極性の環境(いわばエステル基に溶媒和されるような)に存在するサイトでトラップされると考えた。
近似的には、D0で電荷分離の後、D1,D2,・・・,Di とホール移動していく場合、Diでカチオンがトラップされる確率は、p (1-p)i に比例しイオン対間距離の分布は i に依存する。p は、あるサイトがトラップとなる確率である。このモデルに基くとトラップされた Di+ に対する距離分布は
P(ri) = P0 exp[-ri /β]
と与えられる。ここでri は、A-とDi+間の距離で、結合角を考慮した高分子の繰り返し単位の長さravとiの積で
与えられる。βは、
β= - rav / ln(1-p)
である。電子移動反応速度は対間距離の増大とともに次の式のように指数関数的に小さくなる。
kCR = kCR (0) exp[- r / a ]
aの値(この逆数をb値として表す場合も多い)は系に強く依存するが、ここでは剛体溶媒中やアルキル鎖をスペーサーとして用いた場合の値の平均値である1Aを用いた。これらの式を用い電荷分離状態の再結合挙動は下の式で表される。
A(t)∝ΣP(i) exp[-kCR(i) t]
Fig.7(b)の実線は、pとして0.22を用いた場合の計算値である。電荷再結合はナノ秒領域から十時間といった広範囲の時間スケールに及ぶが、おおむね上記の分布と電荷再結合速度の距離依存性を考慮したモデルで実験結果が再現できる。
このような超長寿命イオン対は他のビニルポリマーを用いた場合にも観測され34)、同じβの値を用いて電荷再結合挙動が再現できた。また電子受容体を含まずPVCzだけ吸着させ、比較的強い輝度のレーザー光を用いて二光子イオン化を経由した電荷分離状態を生成することもできる。この場合、初期カチオン-電子分布はTCNBとの電子移動の場合とは異なるが、やはり同じβでカチオンがトラップされることが示された35)。これらの結果から考えると、このβは吸着表面のエステル基周辺の高極性サイトの規則長とビニルポリマーの規則長のマッチング(いわば最小公倍数)によって決定される値であると考えられる。このようなマッチングを最適化設計した表面の作成について現在検討している。
6.まとめと今後の展開
従来複雑であると考えれれてきた高分子系の電子移動ダイナミックスが、比較的簡単なスキームで表現できることを述べた。もちろんいわゆるtrap siteのホール移動ダイナミックスへの影響36)やフェムト秒ダイナミックスなど、基礎的な観点から解明を行う必要が有る点も多数存在する。一方、このような概念を発展させるアプローチもあり、特に超長寿命電荷分離状態の作成などについて、様々な観点から研究を展開している。
文献
1. J. Mort and G. Pfister. in Electronic Properties of Polymers. eds. J. Mort,G. Pfister (Wiley-Interscience, New York.1982) ch.6.
2. M. Yokoyama, S. Shimokihara, A. Matsubara, H. Mikawa: J. Chem. Phys..76(1982)724.
3. Y. Okamoto and A. Itaya, Bull. Chem. Soc. Jpn., 57 (1984) 1626.
4. 板谷 明,「ポリビニルカルバゾール」,ぶんしん出版, (1990).
5. M. Van der Auweraer, F .C. De Schryver , P.M. Borsenberger , H .Bassler, Adv. Mater. 6 (1994) 199.
6. 左貝潤一,「位相共役光学」, 朝倉書店 (1990).
7. R. A. Marcus, N. Sutin, Biochim. Biophys. Acta., 811 (1985) 265.
8. Electron Transfer in Inorganic, Organic and Biological Systems, Advances in Chemistry Series 228, (Eds. J. R . Bolton, N. Mataga, G. McLendon) American Chemical Society (1991).
9. Dynamics and Mechanisms of Photoinduced Electron Transfer and Related Phenomena (Ed. by N.Mataga, T. Okada, H. Masuhara ) Elsevier Amsterdam. (1992).
10. 有機電子移動プロセス (日本化学会編)学会出版センター, (1988).
11. (a) N. Mataga , H. Miyasaka, Prog. React. Kinetics, 19 (1994) 317. (b) N. Mataga, H. Miyasaka, Adv. Chem. Phys., 107 (1999) 431.
12. 増原 宏, レーザー研究, 13 (1985) 722 .
13. T. Ueda, R. Fujisawa, H. Fukumura, A. Itaya, H. Masuhara, J. Phys. Chem., 99 (1995) 3629.
14. K. Watanabe, T. Asahi, H. Masuhara, Chem. Phys. Lett., 233 (1995) 69.
15. H. Miyasaka, T. Moriyama, S. Kotani, R. Muneyasu, A. Itaya, Chem. Phys. Lett., 225 (1994) 315.
16. H. Miyasaka, T. Moriyama, S.R. Khan, A. Itaya in Femtochemistry, Eds. F. C. De Schryver et al. (Wiley- VCH, Weinheim, 2001) p. 335.
17. A. Itaya, T. Kitagawa, T. Moriyama, T. Matsushita, H. Miyasaka, J. Phys. Chem., B 101 (1997) 524.
18. H. Miyasaka, T. Moriyama, T. Ide, A. Itaya, Chem. Phys. Lett., 292 (1998) 339.
19. J. H. Slowik , I. Chen, J. Appl. Phys., 54 (1983) 4467.
20. U. Albrecht, H. Bassler, Chem. Phys. Lett., 235 (1995) 5131.
21. K. Watanabe, T. Asahi , H. Masuhara, J. Phys. Chem., B 101 (1997) 5131.
22. 渡邊一也, 増原 宏, 分光研究, 46 (1997) 229.
23. T. Ikoma, M. Nakai, K. Akiyama, S. Tero-Kubota, T. Ishii, Angew. Chem. Int. Ed., 40 (2001) 3234.
24. H. Miyasaka, T. Moriyama,, A. Itaya, J. Phys. Chem., 100 (1996) 12609.
25. T. Moriyama, K. Monobe, H. Miyasaka, A. Itaya, Chem. Phys. Lett., 275 (1997) 291.
26. H. Miyasaka, T. Moriyama, A. Itaya, J. Phys. Chem., B101 (1997) 10726.
27. 宮坂 博、季刊化学総説44, 超高速化学ダイナミックス, 学会出版センター, (2000) p.189.
28. H. Miyasaka, S. R. Khan, A. Itaya, J. Phys. Chem., A 106 (2002) 2192.
29. (a) T. Asahi, N. Mataga, J. Phys. Chem. ,93 (1989) 6575. (b) T. Asahi, M. Ohkohchi, N. Mataga, J. Phys. Chem., 97 (1993) 13132.
30. A. Tsuchida, A. Nagata, M. Yamamoto, H. Fukui, M. Sawamoto, T. Higashimura, Macromolecules, 28 (1995) 1285.
31. (a) A. Osuka, T. Nagata, F. Kobayashim R. P. Zhang, K. Maruyama, N. Mataga, T. Asahi, T. Ohno, K. Nozaki, Chem. Phys. Lett., 199 (1992)302. (b) A. Osuka, R. P. Zhang, K. Maruyama, T. Ohno, K. Nozaki, Bull. Chem. Soc. Jpn., 66 (1993) 3773.
32. H. Imahori, D. M. Gludi, K. Tamaki, Y. Yoshida, C. Luo, Y. Sakata, S. Fukuzumi, J. Am. Chem. Soc., 123 (2001) 6617
33. (a) S. Kotani, H. Miyasaka, A. Itaya, J. Phys. Chem., 99(1995) 13062. (b) S. Kotani, H. Miyasaka, A. Itaya, J. Phys. Chem., 100(1996)19898.
34. H. Miyasaka, T. Ota, S. Kotani, A. Itaya, Mol. Cryst. Liq. Cryst., 315 (1998) 193.
35. H. Miyasaka, S. Kotani, T. Ota, A. Itaya, Chem. Phys. Lett., 335 (2001) 496.
36. S. R. Khan, K. Ohi, A. Itaya, T. Okada, H. Miyasaka, Phys. Chem. Chem. Phys., 5 (2003) 1003.
![]()