フォトクロミック分子系のレーザー多光子吸収による反応制御
1.フォトクロミック反応
フォトクロミック反応とは、光照射によって分子量の変化を伴わず分子内での化学結合の組み替えなどによって分子の構造変化が起こる過程(光誘起異性化反応)を示す。それぞれの分子量は等しいが結合の形式などが異なる分子は異性体と呼ばれる。異性体 AとBの間の変化(A→B及びB→A)のうち、少なくとも一方が光照射によって進行する場合、フォトクロミズムと呼ばれる[1]。
図1には、フォトクロミック反応の一例を示した。
両異性体の吸収波長、屈折率、双極子モーメントなどの種々の物性はそれぞれに異なる。したがって光照射によって素早くこれら諸物性の変化を伴うフォトクロミック反応は、光メモリーや光スイッチなどのフォトニクスデバイスへの応用といった観点から活発な研究がなされている。 また電子状態の変化によって反応性が変化する系は、光化学反応の最も特徴的な系であり、基礎的な光反応としても興味深く、我々を含めパルスレーザーを用いた機構解明に関する研究も多くなされている[2]。
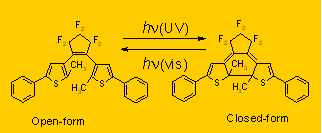
図1.ジアリールエテン誘導体のフォとクロミック反応の一例。
紫外光照射によって、Open-form (開環体)→ Clsoed-form(閉環体)へ、可視光照射によって、Clsoed-form → Open-formのフォトクロミック反応を起こす。
現在までにアゾベンゼン、スピロピラン、スピロオキサジン、フルギドなど数多くのフォトクロミック分子系が提案、開発されてきている。(これらの詳細は、2000年5月に発光されたChemical Reviews(上)に総説が掲載されている。)これらの分子群の中で、ジアリールエテン誘導体は、近年、入江らによって開発されたフォトクロミック分子であり両異性体が熱的に安定であり、繰り返し耐久性にも優れ、両異性化反応が光誘起によって進行するといった特徴を持つ[1]。
フォトクロミック化合物を実際にメモリーのようなデバイスに利用するためには、
(1) 異性体の熱的安定性、
(2 )高い繰り返し耐久性、
(3) 高速応答性、
(4) 高感度、
(5)非破壊読み出しが可能、
などの条件が要求される。フォトクロミック反応は、有限の励起状態寿命の間に発光過程や無輻射過程などと競争して進行する。したがって、本質的に異性化反応速度の大きい系(高速応答性)は、反応収率も大きく(高感度)、結果的に副反応を抑制できる(高耐久性)といった主要な条件を満足する。一方、 (4)の高感度性と矛盾せずに (5)の非破壊読み出しを行うためには、反応の進行を抑制あるいは促進する何らかの外部条件(ゲート)が必要となる。光によるゲートの制御は、波長、光強度等いくつかのパラメーターを系と独立に設定できること、また高速制御も可能であることから有効な方法と考えられる。我々は、ジアリールエテンやフルギド誘導体を対象に、ピコ秒、フェムト秒レーザー分光による反応ダイナミックスやメカニズムの解明、また開環反応を対象に、光強度による反応制御(多光子反応ゲート機能)について研究を行っている[3,4,5]。
現在までの研究の結果では、ピコ秒レーザーを励起光源に用いた場合、定常光照射の場合に比べて、数10倍から1000倍程度の反応効率の促進を達成している。これは、一光子吸収によって生成されたS1状態が、更にもう一光子吸収して生成した高い励起状態(Sn)からの開環反応効率が大きいことによる。
2.多光子制御フォトクロミック反応
一例を述べる。図1に示したジアリールエテン誘導体の1つ(PC1)は、この図にに示すような環開閉フォトクロミック反応を行う。開環体(Open-form)は紫外部にのみ吸収を持つが、閉環体(Closed-form)は可視部にも吸収を持ち青色に着色している。このPC1の開環反応収率(Closed-form→Open-form)は、定常光照射では1.3%と非常に小さい。
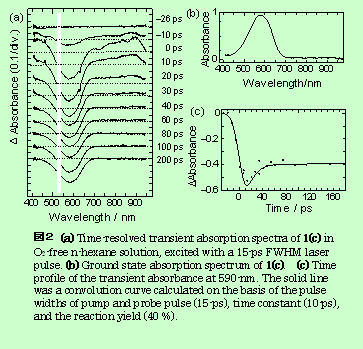
ピコ秒532nmレーザーでPC1閉環体(Closed-form) / n-ヘキサン系を励起した場合、図.2(a)に示す様な過渡吸収スペクトルが得られた。590nm付近の負の吸収シグナルは、図2(b)に示す閉環体の定常状態の吸収スペクトルと対応しており、基底状態の閉環体が励起されたことを示す。励起直後に700nmより長波長に現れる正の吸収は10psの時定数で減衰する。
図2(c)は、590nmの過渡吸光度の時間変化を示す。532nm励起によって基底状態分子が励起状態に上がり、その後減衰する挙動を示す。回復の時定数は10psであり上に述べた正の吸収の減衰時定数と一致することから、700nmより長波長に観測される正の吸収は、励起状態から更に高い励起状態への吸収と同定できる。この一定成分は、開環反応(Closed-form→Open-form)によって開環体が生成した事を示している。この励起光強度の条件では反応収率は40%と見積もられ、定常光照射の場合(1.3%)の30倍以上の増大が観測された。
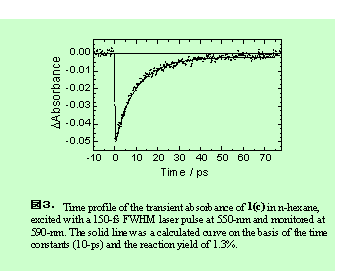
一方、PC1/ n-ヘキサン系を610nmフェムト秒レーザー光で励起し、590nm(基底状態の吸収極大)でモニターしたところ、基底状態のブリーチは12psの時定数で回復した後、開環反応に対応した1%程度の一定成分が残った (図3) 。すなわちフェムト秒レーザー励起の場合は、定常光照射と同程度の反応収率が観測された。なお、励起波長を540 nmとしたときにも同様の結果が得られた。
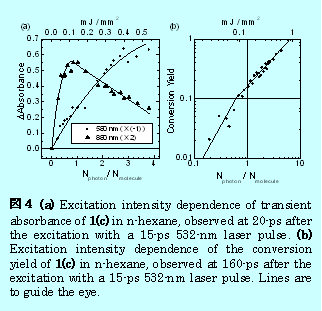
この励起条件に依存した反応挙動の違いを探るため、ピコ秒532nmレーザー光強度を変化させ、過渡吸収スペクトルのの形状に与える影響を検討した。図4にその結果を示す。図4(a)は、励起後20psでの過渡吸収スペクトルに与える励起光強度の影響を示している。横軸は、基底状態の分子数とレーザー光の光子数の比で励起光強度を示した。▲は880nmでモニターした閉環体の励起状態の吸収強度である。励起光強度があまり大きくない領域では、励起光強度の増大とともにこの吸収強度は大きくなるが、更に大きくなるとこの吸収は減少する。一方、基底状態分子の現象に対応した580nmでは、励起光強度の増大とともに単調に吸収強度は増大する。図4(b)には、励起後160ps(反応が終了した時間)での閉環体の負の吸収に対する励起光強度依存性を示した。先述の通りこの吸収強度は光反応によって生成した開環体の量に対応している。
基底状態の分子数とレーザー光の光子数の比が1程度までは、開環体は励起光に対して二光子過程によって生成する。一方それ以上の励起光強度では見た目の傾きは1になっている。強い励起光強度条件では基底状態分子のほとんどが既に励起状態に存在するため、傾きが一に近くなったと考えられる。図4からピコ秒レーザー励起による開環体の生成は、二光子吸収過程が寄与していることがわかった。
一般に、二光子吸収は、①Simultaneous Processと② Stepwise Processの二つの場合に分けられる。①のSimultaneous Processは基底状態からの一光子共鳴吸収帯の有無に関係なく同時に二光子吸収を行う過程であり、パルスの尖頭出力の二乗に比例して吸収確率が増大する。一方、②の過程は一光子吸収で生成した中間体が更にもう一光子吸収する場合に対応する。この場合も単位時間あたりの光子密度が大きいことは重要な因子ではあるが、総光子数が多いことも重要な条件となる。
我々の使用したフェムト秒、及びピコ秒のレーザーはパルス幅が100倍異なる。一方、出力もフェムト秒パルスはピコ秒レーザーの約1/100と小さく、尖頭出力はほぼ同程度となっている。したがってフェムト秒とピコ秒レーザーパルスの単位時間あたりのエネルギー強度(出力/パルス幅)はほぼ同じであり、①の過程の寄与は同程度と見積もられる。更に図4に示される結果は中間体(閉環体の励起状態)が更にもう一光子吸収を行うことによって高い励起状態を経て開環反応が進行することを示しており、主に②のStepwise二光子吸収過程が効率の良い開環反応に寄与していることがわかった。すなわちフェムト秒パルスは、総光子数が少なく基底状態分子のごく一部が励起されるのみであるが、ピコ秒レーザーは総光子数が多く、パルスの前半で照射体積中の大半の分子が励起状態を生成しうる。そのため、基底状態分子の吸収に阻害されることなく励起状態分子がもう1光子を効率よく吸収できることが、高い励起状態の生成を可能とし高効率な開環反応の要因となっている。またこの高い励起状態からの開環反応収率は約30から40%と見積られ、この値が比較的大きいことも効率の良い多光子開環反応過程に寄与している。
これらの結果は、開環反応収率の小さいジアリールエテン誘導体の閉環体に対しては、比較的弱いレーザー光で情報を読み出した場合には非破壊読みだしが可能であるが、一方、若干強いレーザー光を利用した場合には、記録の消去が可能であること(1-color gate機能)を示す。図4に示すように記録消去(開環反応)のための励起条件は、15psのパルス幅のレーザーに対してmJ / mm2程度であるが、同程度のパルス幅のレーザーをμm2に集光できればnJ程度の出力で達成でき、実際のデバイスを想定した場合にも必ずしも非現実的な条件ではない。
また他のジアリールエテン誘導体でもこのような多光子吸収による開環反応の増大が見られている。高い励起状態のポテンシャルエネルギーに関する考察、また1光子吸収による開環及び閉環反応挙動、分子振動に位相を合わせた多光子反応制御を含めて、研究を進展している。[4,5]
1) M. Irie Chem. Rev. 100 (2000) 1685.
2) N. Tamai, H. Miyasaka Chem. Rev. 100(2000) 1875.
3) H. Miyasaka, M. Murakami, A. Itaya, D. Guillaumont, S. Nakamura, M. Irie J. Am. Chem. Soc. 123 (2001) 753.
4) H. Miyasaka, M. Murakami, T. Okada, Y. Nagata, A. Itaya, S. Kobatake, M. Irie, Chem. Phys. Lett., 371 (2003) 40-48.
5) M. Murakami, H. Miyasaka, T. Okada, S. Kobatake, M. Irie, Submitted.